Research
I. Structure, thermodynamics and kinetics of condensed phase interfaces:
Understanding the structure, thermodynamics and kinetics of the interfaces between condensed phase systems is of paramount importance in a wide array of technologically important fields, including crystal growth and nucleation, wetting, material strength and stability, Experimental study is often difficult as such interfaces lie sandwiched between two dense phases. Because
![Snapshot from a molecular dynamics simulation of a premelting at a Al-Pb solid-liquid interface [Phys. Rev. Lett. 110, 096102 (2013)]](/sites/lairdgroup/files/images/research.jpeg)
Snapshot from a molecular dynamics
simulation of a premelting at a Al-Pb solid-liquid interface
[Phys. Rev. Lett. 110, 096102 (2013)]
experimental data is lacking or often incomplete, molecular modeling studies of such systems can offer valuable insight into their detailed molecular phenomenology. Much of our work in this area in the past has focused on the solid-liquid interfaces of simple, one-component model systems. In recent years, our focused has shifted to interfaces in complex systems such as multicomponent systems (e.g., metal alloys), systems with molecular interactions and grain boundaries. For such studies, we combine state of the art molecular simulation techniques and applied statistical mechanics to a variety of realistic and model systems. Recent publications include
- "Thermodynamics and Intrinsic Structure of the Al-Pb Liquid-Liquid Interface: A Molecular-Dynamics Simulation Study", Y. Yang and B.B. Laird, J. Phys. Chem. B (Jim Skinner Festschrift), 118 8373-8380 (2014).
- "Calculation of the interfacial free energy of a binary hard-sphere liquid at a planar hard wall", J.L. Kern and B.B. Laird, J. Chem. Phys., 140, 024703:1-7 (2014).
- "Solid-liquid interfacial premelting", Y. Yang, M. Asta and B.B. Laird, Phys. Rev. Lett. 110, 096102 (2013).
- "Interfacial free energy of a hard-sphere fluid in contact with curved hard surfaces", B.B. Laird, A. Hunter and R.L. Davidchack, Phys. Rev. E, 86 060602:1-5 (2012).
II. Phase-Equilibrium and Transport properties of Novel Solvent Media for Environmentally Beneficial Catalysis:
For the last several years we have been working closely with KUs Center for Environmentally Beneficial Catalysis, an engineering research center whose mission is the development and application of environmentally friendly catalytic processes for industry. Much of our effort has gone into molecular modeling studies to support the development of novel "green" solvent media for catalysis. One class of solvents that is of interest are so called gas-expanded liquids (GXLs), in which a large amount of gas (usually carbon dioxide) is dissolved into a traditional organic solvent, expanding its volume appreciably. The resulting solvents are environmentally beneficial in that they reduce the amount of potentially harmful organic solvents necessary to carry out a particular catalytic reaction. These solvent systems also have enhanced mass transport and reactant solubility properties over the neat organic solvents and over super-critical carbon dioxide. We have been using a number of molecular simulation techniques (Gibbs Ensemble and Grand Canonical Monte Carlo and Molecular Dynamics Simulation) to study the phase-equilibrium, structure and transport properties of these novel solvents.
Recently, we have entered into a new collaboration with the CEBC funded by the National Science Foundation program Networks for Materials Synthesis and Design. This is a highly multidisciplinary and multiscale effort involving several KU research groups: two in Chemical Engineering (Subramaniam and Choudhary), two in Computational Chemistry (Laird and Thompson) and one Organic Chemistry group (Tunge).This highly collaborative effort uses molecular modeling, synthesis, reactor engineering, kinetic studies and life-cycle assessments to develop and optimize the design of industrial catalytic processes for minimal environmental impact. This framework is being applied to two examples of high-volume chemical processes. One is a safer, phosgene-free route to dimethyl carbonate, which is used to manufacture polycarbonates and polyurethanes. The other is a cleaner, more efficient route from butadiene to adipic acid, used to make nylon. The discoveries and methodologies developed for these two chemicals will also find broader impacts for other major chemical processes. Our theoretical/computational modeling will provide a detailed understanding of the molecular processes involved in the complex reactions, which are often inaccessible through experiment alone. For example, in a new synthetic route to DMC, ethylene is oxidized to ethylene oxide inside a catalytic metal-exchanged amorphous silica mesopore. Molecular simulation allows us to measure the nano-confined reactant system more easily than typical experiments allow. Generally, we are able to study the phase behavior, structure, and transport properties of these novel reaction environments.
- "Prediction of phase equilibria and transport properties in carbon-dioxide expanded solvents by molecular simulation", Y. Houndonougbo, B.B.Laird and K. Kuczera, Mol. Simul. 33, 861-869 (2007).
- "Transport properties in carbon dioxide-expanded acetonitrile from molecular-dynamics simulations", Y. Houndonougbo, B.B. Laird and K. Kuczera", J. Chem. Phys., 126 074507 (8 pages) (2007).
- "Phase Equilibria in Carbon Dioxide Expanded Solvents: Experiments and Molecular Simulations", Y, Houndonougbo, J. Hong, B. Rajagopalan, K. Wong, K. Kuczera, B. Subramaniam, and B.B. Laird, J. Phys. Chem. B 110, 13195-13202 (2006).
- "Monte Carlo Simulations of carbon-dioxide-expanded Acetonitrile", Y. Houndonougbo, J-X Guo, G.H. Lushington and B. Laird, Mol. Phys. 104, 2955--2961 (2006).
III. Development of new algorithms for molecular modeling
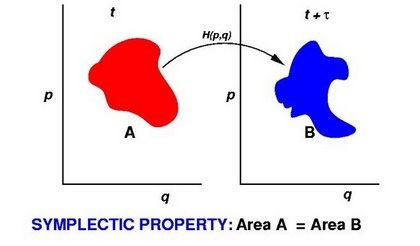
Molecular dynamics computer simulation has become an invaluable tool in chemistry, chemical engineering, physics,materials science and biology; however, its uses are still limited by the relatively small system sizes and short time scales that can be simulated atpresent. Progress in this area therefore comes from advances in computer technology and in the development of efficient and stable algorithms. The latter is the goal of an multidisciplinary project involving the Laird group in collaboration with Prof. Ben Leimkuhler, an applied mathematician at the University of Edinburgh.
One central property of Hamiltonian systems used in molecular dynamics simulations is that it is sympletic, that is, the sum of oriented areas in phase space is a conserved quantity. A well- known consequence of this property is the conservation of phase space volume in Hamiltonian dynamics - otherwise known as Liouville's theorem. It has been shown that stable numerical integrators for molecular dynamics can be constructed by requiring that these approximate integrators are themselves sympletic. We have applied symplectic numerical integration techniques to a number of problems in molecular simulation ranging from rigid body motion to extended Hamiltonian methods for constant-temperature and/or constant pressure molecular dynamics. Representative publications are
- "Generating Generalized Distributions from Dynamical Simulation", E.J. Barth, B.B. Laird and B.J. Leimkuhler, J.Chem. Phys., J. Chem. Phys. 118 5759-5767 (2003)
- "Generalized dynamical thermostatting", B.B. Laird and B.J. Leimkuhler, Phys. Rev. E 68, 016704 (2003).
- "Constant-temperature molecular-dynamics algorithms for mixed hard-core/continuous potentials" , Y.A. Houndonougbo and B.B. Laird, J. Chem. Phys. 117, 1001 ( 2002).
- "Symplectic algorithm for constant-pressure molecular-dynamics using a Nose-Poincare thermostat" J.B. Sturgeon and B.B. Laird, J. Chem. Phys. 112 , 3474--3482 (2000)
- "Molecular dynamics algorithms for mixed hard-core/continuous potentials'' Y.A. Houndonougbo, B.B. Laird and B.J. Leimkuhler, Mol. Phys., 98 309--316 (1999).
- "The Nose-Poincare method for constant temperature molecular dynamics'', S.D. Bond, B.J. Leimkuhler and B.B. Laird, J. Comp. Phys. 151, 114-134 (1999).